Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
The transient receptor potential Ankyrin 1 signaling pathway in hypoxic preconditioning
Time:2024-09-16
Number:11920
Author Affiliations

Conditioning Medicine 2023. 6(6): 197-207.
Abstract
Hypoxic preconditioning has the potential to represent a valuable intervention to trigger and amplify the body’s endogenous protection against stress. In contrast with classical ischemic preconditioning, hypoxic preconditioning is a non-invasive procedure that can be applied in multiple ways, such as by breathing gas mixtures with variable oxygen concentrations or by varying the body’s oxygen consumption via physical exercise. As far as the cardiovascular system is concerned, such interventions target multiple sub-systems, triggering various systemic responses and causing elevated cellular and molecular changes. Such changes interact collectively to lead to a cardioprotection outcome. Among the potentially involved cellular and molecular changes, here we focus on the pathway that originates from the modulation of CA2+ entry into the cardiac myocytes via the transient receptor potential Ankyrin 1 (TRPA1) channel. Being hypoxia-sensitive because it is strictly associated with hypoxia-inducible factors, this pathway may represent an ideal link between hypoxic preconditioning and the insurgence of cardioprotection. Here, we briefly review the mechanisms of action of TRPA1 in the cardiovascular system that could be recruited by TRPA1-mediated extracellular Ca2+ entry during hypoxic preconditioning. The availability of dietary and synthetic TRPA1 agonists that may potentially activate TRPA1 for cardioprotective purposes may open new frontiers to design novel therapeutic approaches for preconditioning medicine.
Keywords: hypoxic preconditioning, TRPA1, calcium entry, cardioprotection, hypoxia-inducible factor, cardiovascular system
Abstract
Hypoxic preconditioning has the potential to represent a valuable intervention to trigger and amplify the body’s endogenous protection against stress. In contrast with classical ischemic preconditioning, hypoxic preconditioning is a non-invasive procedure that can be applied in multiple ways, such as by breathing gas mixtures with variable oxygen concentrations or by varying the body’s oxygen consumption via physical exercise. As far as the cardiovascular system is concerned, such interventions target multiple sub-systems, triggering various systemic responses and causing elevated cellular and molecular changes. Such changes interact collectively to lead to a cardioprotection outcome. Among the potentially involved cellular and molecular changes, here we focus on the pathway that originates from the modulation of CA2+ entry into the cardiac myocytes via the transient receptor potential Ankyrin 1 (TRPA1) channel. Being hypoxia-sensitive because it is strictly associated with hypoxia-inducible factors, this pathway may represent an ideal link between hypoxic preconditioning and the insurgence of cardioprotection. Here, we briefly review the mechanisms of action of TRPA1 in the cardiovascular system that could be recruited by TRPA1-mediated extracellular Ca2+ entry during hypoxic preconditioning. The availability of dietary and synthetic TRPA1 agonists that may potentially activate TRPA1 for cardioprotective purposes may open new frontiers to design novel therapeutic approaches for preconditioning medicine.
Keywords: hypoxic preconditioning, TRPA1, calcium entry, cardioprotection, hypoxia-inducible factor, cardiovascular system
Highlights
Hypoxic preconditioning is an emergent procedure that can be applied actively (e.g., physical exercise) or passively (e.g., breathing gas mixtures with variable oxygen levels) to raise cardioprotection. A multitude of subsystems, systemic responses, as well as cellular and molecular changes mediate the translation of hypoxic preconditioning into protective signals. Among these, the modulation of calcium entry via the interaction between the factors induced by hypoxia and the hypoxia-sensitive transient receptor potential ankyrin 1 (TRPA1) channel may be a key driver of hypoxic preconditioning in the cardiovascular system. The availability of dietary and synthetic TRPA1 agonists may open a new frontier for designing therapeutic approaches to preconditioning medicine.
Introduction
Preconditioning is broadly defined as the situation that occurs when a tissue, an organ, or even the entire body is exposed repeatedly to sublethal stresses or stimuli to gain resilience against potentially lethal stresses or stimuli (Holcombe and Weavers, 2022). The root of this phenomenon is hormesis, a complex event first examined in toxicologic studies: while a high-dose contact with a harmful agent is accompanied by toxic effects, a low-dose contact with the same agent may call for beneficial effects instead (Mattson, 2008). This biphasic dose response to environmental agents occurs in several instances. For example, myocardial ischemic preconditioning (IschPrec), first discovered in 1986 (Reimer et al., 1986), is a representative example of preconditioning medicine. In myocardial IschPrec, previous exposures to brief periods of vascular occlusion make the heart resistant to the subsequent effects of major ischemic events. As no known pharmacological or procedural interventions are yet recognized to be as efficient, IschPrec is still the most potent maneuver to improve cardioprotection in experimental models. However, translating IschPrec into clinical practice has proved disappointing, despite the identification of the main mechanisms that confer protection, mainly endothelial nitric oxide (eNOS)-protein kinase G (PKG), reperfusion injury salvage kinase (RISK), survivor activating factor enhancement (SAFE), ATP-sensitive potassium ( K+) channels, and calcium (Ca2+) overload (Comita et al., 2021). One reason for this poor clinical translation is perhaps the complex nature of IschPrec, as it involves not only the contracting cardiomyocytes but also the endothelium, the microcirculation, the immune system, cardiac remodeling, the coagulation cascade, and, more recently, the onset of inflammation (Algoet et al., 2023). Another reason is the invasive nature of classical IschPrec. To overcome the latter concern, non-invasive alternatives such as remote conditioning, post-conditioning, delayed IschPrec, perhaps physical exercise, and hypoxic preconditioning (HypPrec) have been occasionally proposed.
Here, we briefly review the mechanisms induced by HypPrec, which involves a novel key pathway that can be activated by hypoxia and targets Ca2+-sensitive cardioprotective mechanisms, the transient receptor potential (TRP) ankyrin 1 (TRPA1) pathway. To this purpose, we conducted a comprehensive literature search across PubMed, Google Scholar, and Web of Science using the following keywords: TRPA1, hypoxia, hypoxic preconditioning, cardioprotection, heart, cardiovascular disorders, cardiomyocytes, endothelial cells, and cardiac fibroblasts. The present review is not intended as a systematic review that follows the PRISMA guidelines, but it nevertheless suggests that TRPA1 signaling could be activated during HypPrec and may stand out as a potential target to design novel therapeutic approaches based on preconditioning medicine.
Hypoxic preconditioning
HypPrec today is emerging as a powerful approach to raise protection in virtually all body organs, including the cardiovascular system. HypPrec can be administered in multiple ways by exposing experimental animals and human subjects to high- or low-frequency intermittent hypoxia, continuous hypoxia for sustained periods as during sojourns at high altitude, or even by alternating hypoxia and hyperoxia (Burtscher et al., 2023). Remarkably, all these interventions are noninvasive and may also be administered to passive subjects, such as unconscious or paralyzed patients.
These interventions trigger several systemic responses in virtually every body compartment to accomplish their effects. Such responses, in turn, trigger cellular and molecular responses that collectively stimulate the processes leading to HypPrec (Burtscher et al., 2023). Table 1 reports some of the major targets for HypPrec related to the insurgence of protection in the cardiopulmonary system.
.png)
HypPrec represents the culmination of a complex interaction across several sub-systems, each of which independently triggers multiple systemic responses that affect many overlapping signaling pathways. Most, but not all, of the signaling pathways affected by HypPrec may originate from the intracellular accumulation of hypoxia-inducible factors (HIF) that act as cell and body oxygen (O2) sensors. Remarkably, this orchestrator of the body’s response to hypoxia responds to both a lack of oxygen (O2) and excess O2 (Mancardi et al., 2022). Thus, it is difficult to believe that HIF is the only mechanism underlying the complex response to O2 variations. Clearly, many molecular pathways interact to yield such an extensive outcome, HypPrec, which occurs in virtually all systems in the body in response to major stress.
TRPA1 and its effect on the protection of the cardiovascular system
TRPA1: biophysics and gating
TRPA1 is a unique member of the TRP Ankyrin (TRPA) subfamily of non-selective cation channels in mammals (Talavera et al., 2020). The eponymous feature of TRPA1 is the presence of sixteen ankyrin repeats domain (ARD) within the NH2 terminus of the protein channel, which regulates TRPA1 interaction with protein partners and TRPA1 activation by a variety of stimuli (Talavera et al., 2020; Thakore et al., 2020). TRPA1 is widely expressed in sensory neurons and non-neuronal cells (Negri et al., 2019; Talavera et al., 2020; Thakore et al., 2020; Liu et al., 2021; Oguri et al., 2021; Berra-Romani et al., 2023; McGarr et al., 2023), and modulates cellular activity by mediating an inward Na+ and Ca2+ current that depolarizes the membrane, thereby activating voltage-gated channels in excitable cells. In addition, the ensuing increase in intracellular Ca2+ concentration ([Ca2+]i) can recruit a plethora of Ca2+-dependent signaling pathways that confer TRPA1 the ability to modulate diverse functions in different cell types (Talavera et al., 2020; Thakore et al., 2020).
The fractional Ca2+ current carried by TRPA1 is relatively high (~17%) as compared to other TRP channels and can even be increased to ~23% by agonist-dependent modulation of the ion-conduction pathway (Gees et al., 2010; Moccia and Montagna, 2023). In this view, TRPA1 is a remarkable example of a polymodal TRP channel being able to integrate chemical, physical, and thermal stimuli (Talavera et al., 2020; Thakore et al., 2020; Moccia and Montagna, 2023). TRPA1 is also a temperature-sensitive channel that can be activated by both noxious heat (>43°C) and noxious cold (<12°C) (Moparthi et al., 2016; Vandewauw et al., 2018; Moparthi et al., 2022), although its role in the cold sensation by thermal Aδ fibers is still controversial (Zhang et al., 2022). In addition, TRPA1 is sensitive to many electrophilic and non-electrophilic agonists that covalently or non-covalently, respectively, modulate TRPA1 activation (Talavera et al., 2020; Thakore et al., 2020). Electrophilic activators (such as allicin, allyl isothiocyanate or AITC, and cinnamaldehyde) interact with the thiol (-SH) group of cysteine (Cys)621, Cys641, and Cys665 that are located within the NH2 terminus between the last ARD and the first transmembrane α-helix (Michlig et al., 2016; Talavera et al., 2020; Alvarado et al., 2021; Moccia and Montagna, 2023). Sulfhydryl-reacting compounds that activate TRPA1 include many irritant compounds, such as acrolein, diesel exhaust, and anesthetics (Talavera et al., 2020; Alvarado et al., 2021; Moccia and Montagna, 2023). Covalent modifications of specific Cys residues also underlie TRPA1 sensitivity to reactive oxygen species (ROS) and reactive nitrogen species (RNS), which are regarded as endogenous agonists of TRPA1 (Talavera et al., 2020; Alvarado et al., 2021; Moccia and Montagna, 2023). For instance, TRPA1 can be directly gated by hydrogen peroxide (H2O2) (Bessac et al., 2008; Faris et al., 2023), hydrogen sulfide (H2S) (Kimura, 2024), nitric oxide (NO) (Miyamoto et al., 2009), and peroxynitrite (ONOO-) (Andersson et al., 2015). A growing body of evidence suggests that the lipid peroxidation product 4-hydroxynonenal (4-HNE) is the most widespread endogenous agonist of TRPA1 in mammalian cells (Andersson et al., 2008; Talavera et al., 2020; Alvarado et al., 2021; Moccia and Montagna, 2023). Moreover, TRPA1-mediated inward currents can also be induced by two similar peroxidation products, 4-hydroxyhexenal and 4-oxo-nonenal (Andersson et al., 2008). Finally, TRPA1 activation is also sensitive to intracellular acidification (Riva et al., 2018), depletion of phosphatidylinositol-4,5-bisphosphate (Kim et al., 2008), and changes in intra- and extra-cellular [Ca2+] (Jordt et al., 2004; Wang et al., 2008). Therefore, TRPA1 is a highly versatile channel that integrates multiple cues from the surrounding environment, thereby setting the most appropriate cellular response in motion.
TRPA1 in cardiovascular protection
The pharmacological modulation of TRP channels, including by TRP vanilloid 1 (TRPV1), TRP melastatin 8 (TRPM8), and TRPA1, is coming of age as a novel strategy that protects against a growing number of cardiovascular disorders, including ischemia/reperfusion injury, heart failure, atherosclerosis, hypertension, and critical limb ischemia (Moran, 2018; Moccia et al., 2019; Wang et al., 2019; Negri et al., 2020; Szabados et al., 2020; Moccia et al., 2022a; Moccia et al., 2022b; Bao et al., 2023; Jesus et al., 2023). Understanding which of the multiple cellular components of the cardiovascular system are endowed with TRPA1 is an essential step to designing an effective multitarget strategy based on HypPrec (Davidson et al., 2019). TRPA1 is widely expressed in the cardiovascular system (Wang et al., 2019), especially in vascular endothelial cells (Sullivan et al., 2015; Thakore et al., 2021; Berra-Romani et al., 2023; McGarr et al., 2023), cardiac myocytes (Andrei et al., 2016; Andrei et al., 2019; Conklin et al., 2019), fibroblasts (Oguri et al., 2021), and intracardiac nociceptive fibers (Hoebart et al., 2021; Wang et al., 2022). Although a recent investigation has raised doubt against TRPA1 (and TRPV1) expression in cardiac myocytes (Hoebart et al., 2021), this discrepancy could be due to the different sensitivities of the anti-TRPA1 antibodies (Bao et al., 2023), some of which fail to detect TRPA1 protein either in Western blot or immunocytochemistry assays (Virk et al., 2019), or inappropriate signal-to-noise ratio (Bao et al., 2023). Conversely, TRPA1 is expressed at the mRNA level (Bugert and Kluter, 2006) but not as a protein in platelets (Albarran et al., 2013), while its expression in vascular smooth muscle cells (VSMCs) has never been reported.
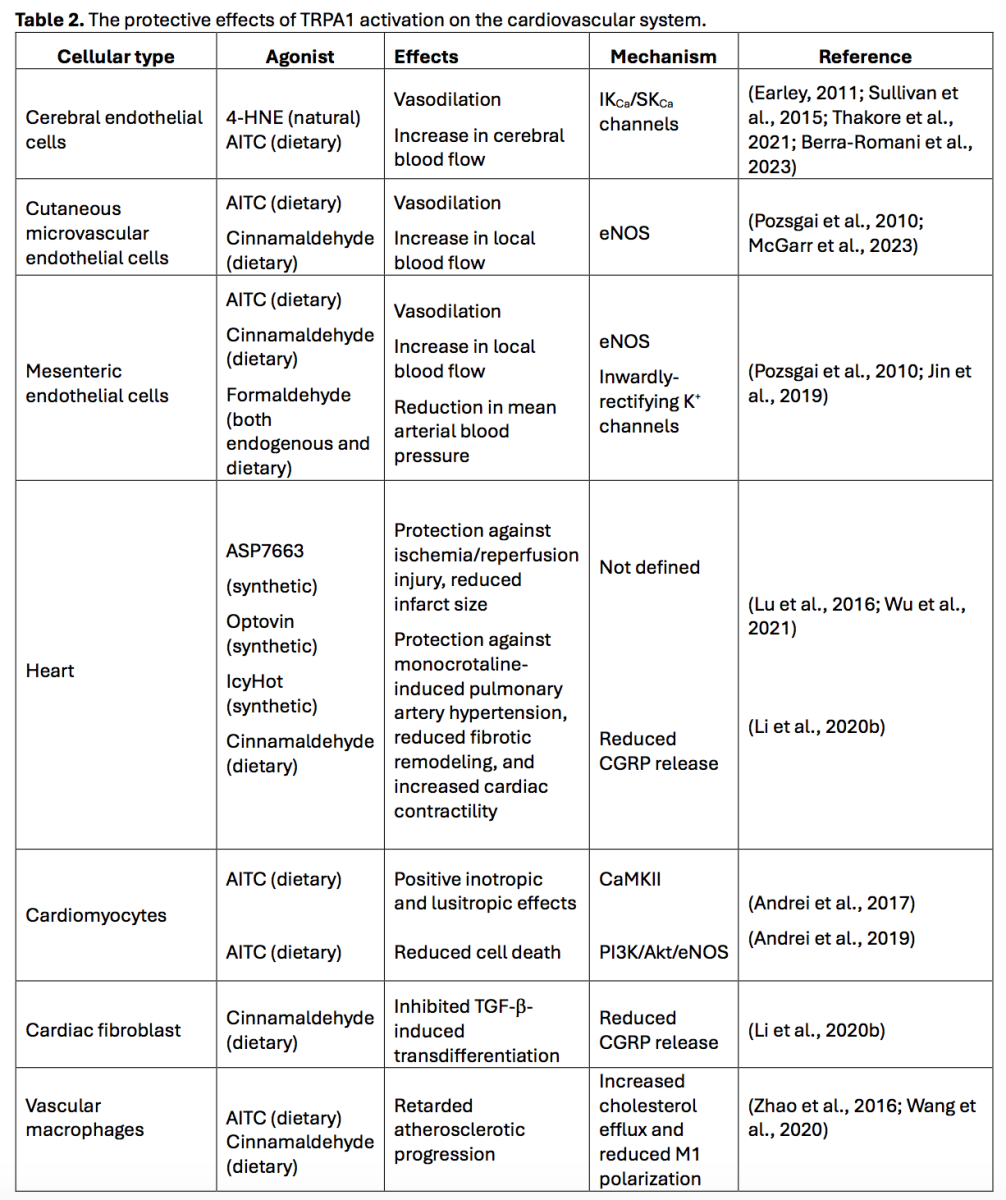
TRPA1-mediated Ca2+ entry recruits vasodilating pathways in vascular endothelial cells
The functional expression of endothelial TRPA1 channels has been reported in several vascular regions (Table 2), including brain (Sullivan et al., 2015; Thakore et al., 2021; Berra-Romani et al., 2023), cutaneous (Pozsgai et al., 2010; McGarr et al., 2023), and mesenteric districts (Pozsgai et al., 2010; Jin et al., 2019), where TRPA1 activation leads to vasodilation and increased local blood flow (Gao et al., 2020; Thakore et al., 2020; Alvarado et al., 2021; Moccia et al., 2022a). TRPA1-mediated Ca2+ entry can recruit at least two distinct endothelium-dependent vasorelaxant pathways (Figure 1), i.e., eNOS and intermediate- and small-conductance Ca2+-activated K+ channels (IKCa and SKCa, respectively) (Earley et al., 2009; Gao et al., 2020; Thakore et al., 2020; Alvarado et al., 2021; Negri et al., 2021; Moccia et al., 2022a). The eNOS and IKCa/SKCa channels can act in synergy to induce a local NO-dependent increase in blood flow at the capillary level, which then spreads to upstream arterioles through IKCa/SKCa-mediated endothelium-dependent hyperpolarization (EDH) to steal blood from inactive tissue areas (Pozsgai et al., 2010; Earley, 2011; Sullivan et al., 2015; Guerra et al., 2018; Thakore et al., 2021; Moccia et al., 2022a; McGarr et al., 2023). It has been reported that cinnamaldehyde exerts endothelium-independent vasorelaxation in ex vivo aortic rings (Yanaga et al., 2006). However, high doses of cinnamaldehydes can inhibit the voltage-gated L-type Ca2+ channels that are expressed in VSMCs (Alvarez-Collazo et al., 2014), which might explain the endothelium- and TRPA1-independent vasorelaxing effect of this compound (Talavera et al., 2020).
In a new window | Download PPT
Figure 1. TRPA1-mediated Ca2+ entry recruits Ca2+-dependent vasorelaxant pathways in proximity to the vascular wall. Extracellular Ca2+ entry through TRPA1 can stimulate eNOS activity, resulting in robust NO release. In addition, TRPA1-mediated Ca2+ signals can activate the small- and intermediated-conductance Ca2+-dependent K+ channels, SKCa and IKCa, which causes endothelium-dependent hyperpolarization (EDH). EDH is then electrotonically propagated to adjacent vascular smooth muscle cells (VSMCs) through myo-endothelial gap junctions, thereby causing VSMC hyperpolarization and relaxation. An alternative mechanism by which TRPA1 activation can induce vasorelaxation is by stimulating the release of NO and CGRP (as well as substance P) from the sensory fibers in close contact with resistance vessels.
Vasodilation can also be induced by the pharmacological activation of TRPA1 expressed on nociceptive neurons of dorsal root ganglion (Gao et al., 2020), which causes peripheral vasodilation through the release of the vasorelaxant neuropeptides, calcitonin gene-related peptide (CGRP) and substance P, and of the gasotransmitter NO (Kunkler et al., 2011; Pozsgai et al., 2012; Aubdool et al., 2016; Hajna et al., 2016). Endothelial TRPA1 channels may also stimulate angiogenesis in the corneal stroma and prostate cancer (Negri et al., 2019) while negatively regulating neovessel formation in coronary circulation (Li et al., 2020a). The dual role of TRPA1 in angiogenesis could be explained by its selective coupling to distinct Ca2+-dependent pathways in different vascular beds, which is a typical feature of many Ca2+-permeable pathways in the vascular endothelium (Moccia et al., 2023a).
TRPA1-mediated Ca2+ entry activates cardioprotective pathways in cardiomyocytes
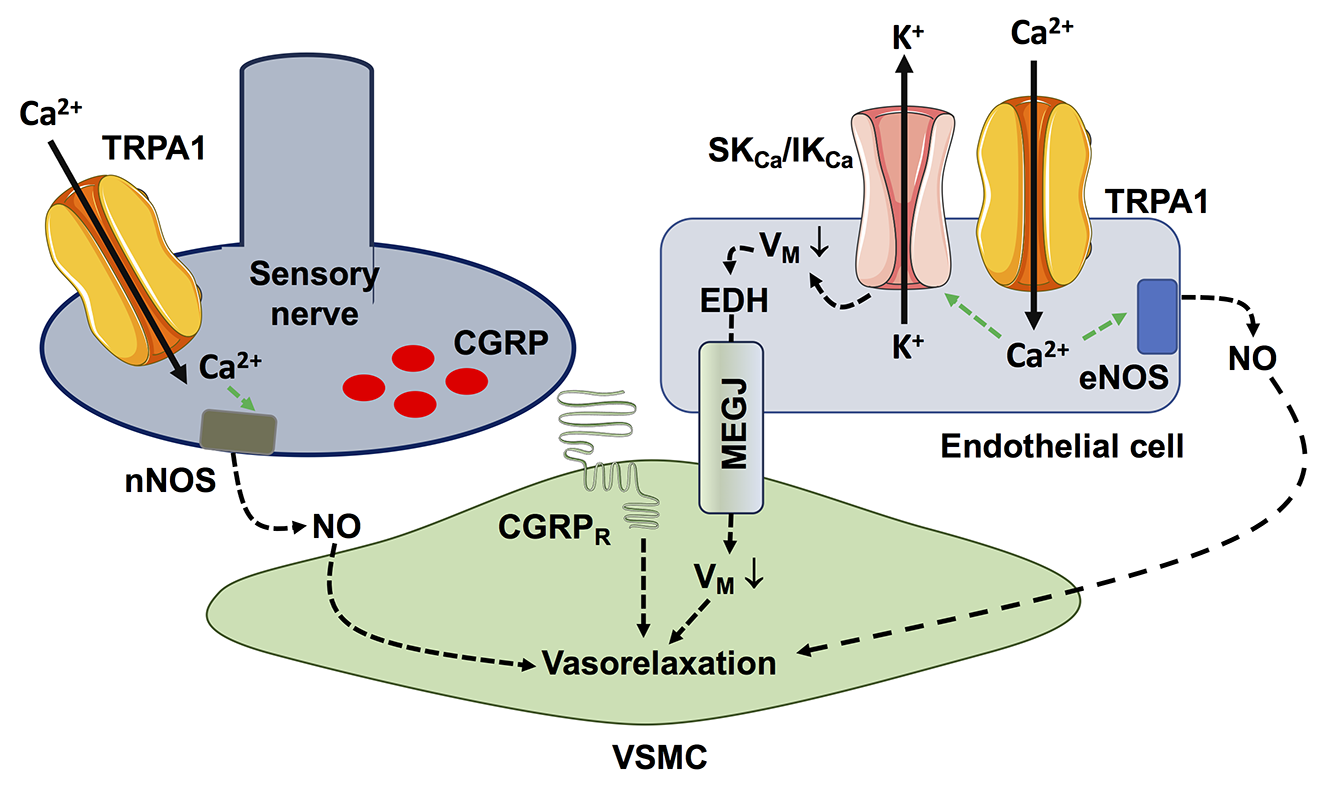
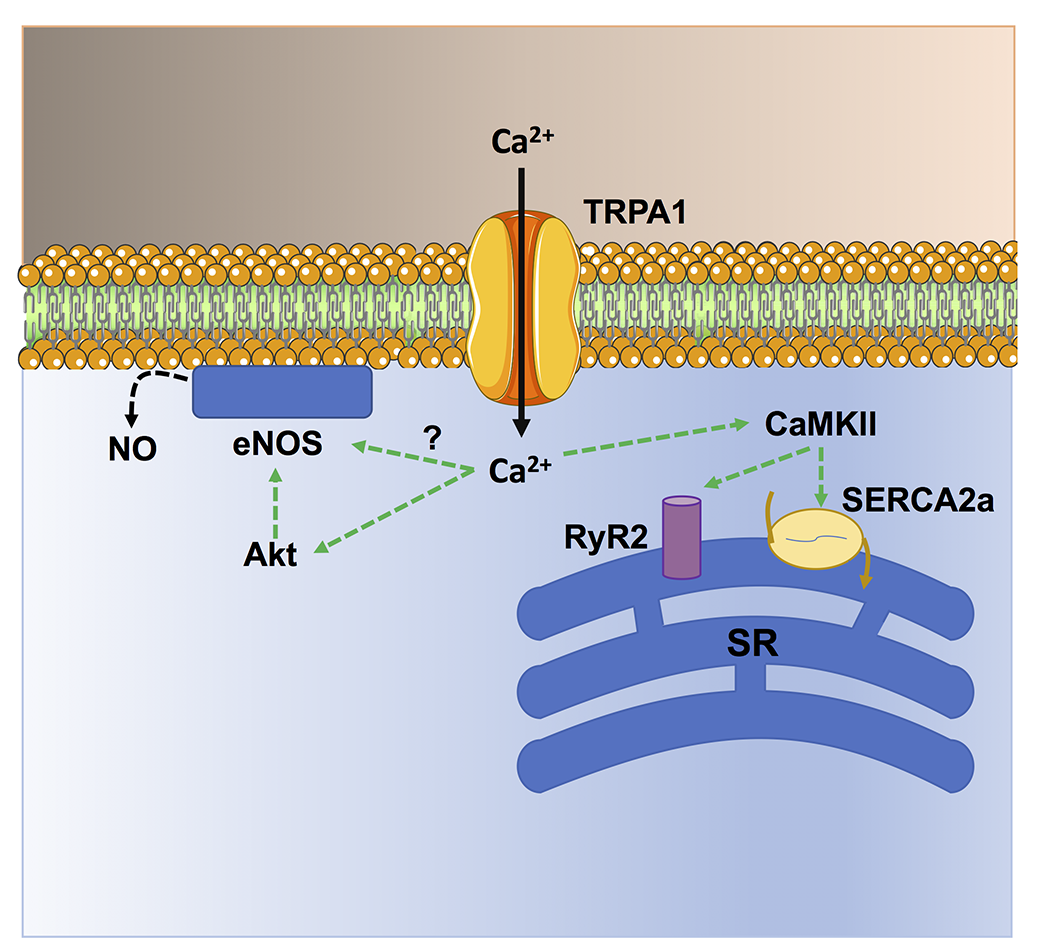
Several studies have shown that TRPA1 activation in the heart is cardioprotective against ischemia/reperfusion injury (Lu et al., 2016; Andrei et al., 2019; Alizadehasl et al., 2024) and prevents myocardial fibrosis (Li et al., 2020b; Ma and Wang, 2022). An early study showed that the pharmacological stimulation of TRPA1 prior to ischemia significantly reduced the infarct size in a rat model of acute myocardial infarction (Lu et al., 2016). Similarly, topical application of the analgesic cream, IcyHot, prior to myocardial ischemia also reduced the infarct size in a TRPA1-dependent manner (Wu et al., 2021). IcyHot was found to release methyl salicylate into the bloodstream during reperfusion, thereby triggering remote cardioprotection by activating TRPA1-dependent cardioprotective pathways (Wu et al., 2021). In vitro studies revealed that TRPA1-mediated Ca2+ entry can both enhance cardiomyocyte contraction and prevent cardiomyocyte cell death (Figure 2) (Alizadehasl et al., 2024). In accord, TRPA1 can induce a positive inotropic and lusitropic effect by engaging Ca2+/calmodulin-dependent protein kinase II (CaMKII) (Andrei et al., 2017), which has long been known to stimulate the Sarcoplasmic Reticulum Ca2+ cycling (Figure 3) (Maione et al., 2020; Reyes Gaido et al., 2023). In addition, TRPA1 stimulation can prevent ischemia-induced cardiomyocyte cell death by recruiting the protein kinase B (Akt)/eNOS signaling pathway (Andrei et al., 2019). It should, however, be pointed out that TRPA1 is also sensitive to ROS, such as H2O2, and aldehydes (Conklin et al., 2019) that can be produced upon reperfusion (Wang et al., 2022). Therefore, the pharmacological activation of TRPA1 prior to inducing ischemia recruits Ca2+-dependent cardioprotective pathways that overcome the detrimental effects of Ca2+ overload at the time of reperfusion (Santulli et al., 2015; Rocca et al., 2023). A major hurdle of these studies consists in the assessment of cardiomyocyte cell death by the lactate dehydrogenase assay (Andrei et al., 2019; Alizadehasl et al., 2024), which reflects membrane damage without discriminating the multiple modes of cell death (Kumar et al., 2018). The mechanisms responsible for cardiac injury during reperfusion do not include only apoptosis and necrosis but also necroptosis, ferroptosis, and pyroptosis (Davidson et al., 2020; Xiang et al., 2024). Future work should explore if TRPA1 activation protects against specific modes of cell death or if it results in global cardioprotective effects. Preliminary evidence suggests that TRPA1-mediated Ca2+ entry reduces the expression of the Ca2+-binding inflammatory protein, S100A8, thereby reducing pyroptosis in rat primary cardiomyocytes (Wang et al., 2023a). Interestingly, Lu et al. (2016) demonstrated that the selective, photosensitive TRPA1 agonist, optovin (Kokel et al., 2013), exerts a strong cardioprotective effect against ischemia/reperfusion injury. Furthermore, optovin has been shown to stimulate cardiomyocyte pacing in both zebrafish hearts in vivo and human stem cell-derived cardiomyocytes in vitro (Lam et al., 2017). Optovin primarily targets the reactive cysteine residues that confer TRPA1 sensitivity to oxidative stress (Trevisani et al., 2007; Kokel et al., 2013; Moccia and Montagna, 2023). However, optovin-induced cysteine modifications are reversible (Kokel et al., 2013), while those induced by ROS and aldehydes are more resistant to endogenous antioxidant systems (Lu et al., 2016). The cardioprotective role of TRPA1 is further supported by the increased cardiovascular risk in patients treated with classical pain relievers that can block TRPA1 activation, such as cyclooxygenase-2 inhibitors or some nonsteroidal anti-inflammatory drugs. during the perioperative period (Lu et al., 2016).
In a new window | Download PPT
Figure 2. TRPA1-mediated Ca2+ entry stimulates Ca2+-dependent cardioprotective pathways in cardiac myocytes. TRPA1-mediated Ca2+ entry stimulates CaMKII, which in turn phosphorylates sarcoendoplasmic reticulum Ca2+-ATPase 2a (SERCA) and type 2 ryanodine receptors (RyR2) to, respectively, sequester cytosolic Ca2+ more avidly (lusitropic effect) and release sarcoplasmic reticulum (SR) Ca2+ more efficiently (inotropic effect). In addition, TRPA1-mediated Ca2+ signals stimulate NO release by inducing Akt-dependent eNOS phosphorylation and, possibly, via direct eNOS activation.
In a new window | Download PPT
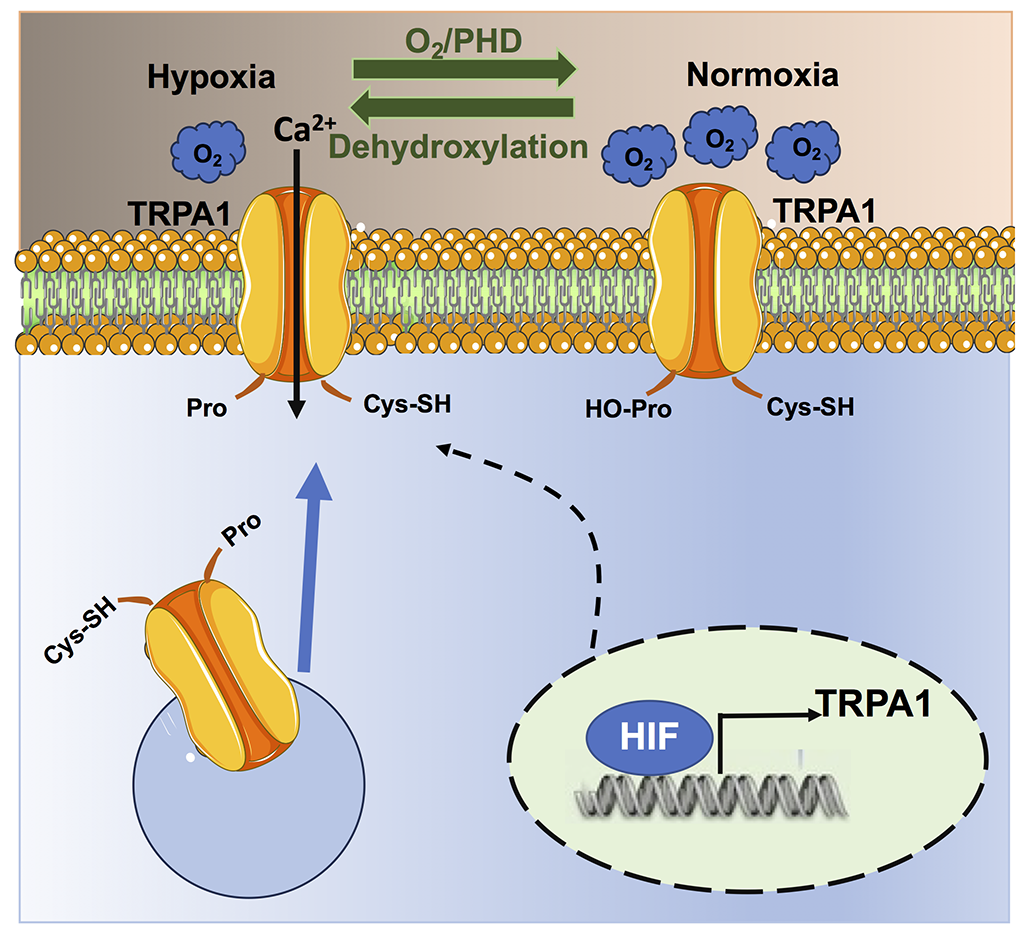
Figure 3. Hypoxia regulates TRPA1 signalling. Under normoxic conditions, PHDs hydroxylate Pro354 on the NH2-terminal ARD domain of the TRPA1 protein, thereby inhibiting the channel and preventing basal Ca2+ entry. The drop in local %O2 under hypoxia inhibits PHD activity, which results in Pro354 dehydroxylation and TRPA1 activation. In addition, hypoxia can lead to the insertion of non-hydroxylated TRPA1 proteins on the plasma membrane, which also results in the increase of basal TRPA1-mediated Ca2+ entry. Finally, hypoxia can increase the expression of TRPA1 protein as the TRPA1 gene contains a hypoxia-sensitive element that is bound by HIF-1.
TRPA1-mediated Ca2+ entry prevents myocardial fibrosis in cardiac fibroblasts
An additional mechanism by which TRPA1 activation can induce cardioprotection is by interfering with cardiac fibrosis. Cardiac fibroblasts are critical determinants of maladaptive ventricular remodeling after an ischemic event (Ma et al., 2017). Intracellular Ca2+ signaling is also involved in cellular differentiation by engaging several Ca2+-dependent transcription factors that regulate cellular fate (Moccia et al., 2015; Maione et al., 2022; Moccia et al., 2023c). Cardiac fibroblasts undergo programmed conversion into myofibroblasts after cardiac injury, thereby releasing a greater amount of extracellular matrix components and favoring cardiac fibrosis (Ma et al., 2017). A recent study showed that the genetic knockout of TRPA1 accelerated aging-induced myocardial fibrosis, ventricular dilation, and cardiac dysfunction, which further hints at TRPA1 as a suitable molecular target to promote cardioprotection (Ma and Wang, 2022). Consistent with this, Li et al. (2020a) showed that the pharmacological stimulation of TRPA1 can also ameliorate myocardial fibrosis by stimulating CGRP release from cardiac fibroblasts both in vitro and in a mouse model of cardiac fibrosis (Li et al., 2020b). CGRP can then autocrinally act by preventing transforming growth factor-β (TGF-β)-induced cardiac fibroblast trans-differentiation via nuclear factor kappa-light-chain-enhancer of activated B cell activation. This mechanism could potentially be enhanced by TRPA1-regulated CGRP release from intracardiac nociceptive fibers (Hoebart et al., 2021; Wang et al., 2022), which also supports cardiomyocyte survival during ischemia/reperfusion injury (Hoebart et al., 2023). Other studies confirmed that TRPA1 deficiency can accelerate cardiac fibrosis in other cardiovascular disorders, such as dilated cardiomyopathy (Wang et al., 2023a). It should, however, be noted that other studies provided contrasting results showing that TRPA1 activation causes TGF-β-induced cardiac fibroblast trans-differentiation both in vitro and in vivo (Wang et al., 2018; Li et al., 2019; Naert et al., 2021; Wang et al., 2023b). It has been suggested that the dual role of TRPA1 in cardiac fibrosis could depend either on the signaling pathway that triggers scar formation or on the stage of myocardial fibrosis (Gao et al., 2020). In addition, the role of TRPA1 signaling in immune cells should be considered (Naert et al., 2021). TRPA1 could drive macrophages towards the M2 phenotype, favoring the infiltration of M2 macrophages in the heart, thereby aggravating pressure-overload induced cardiac hypertrophy and fibrosis (Wang et al., 2018). Therefore, the extent of macrophage polarization and macrophage recruitment into the cardiac interstitium could also determine whether TRPA1 activation primarily protects or exacerbates cardiac fibrosis.
TRPA1-mediated Ca2+ entry in macrophages inhibits atherosclerosis
TRPA1 activation in vascular macrophages, which are the major immune cell population in atherosclerotic lesions (Xu et al., 2019), counteracts M1 polarization, slowing atherosclerosis progression (Gao et al., 2020). TRPA1 expression was found to be increased in macrophage foam cells from the aorta of apolipoprotein E-deficient mice (Zhao et al., 2016). TRPA1 stimulation with AITC retarded atherosclerosis progression and reduced the inflammatory response in atherosclerotic mice. Conversely, either the pharmacological blockade of TRPA1 activity with HC-030031 or the genetic ablation of the TRPA1 protein worsened atherosclerotic lesions, hyperlipidemia, and systemic inflammation (Zhao et al., 2016). A subsequent study confirmed these findings, showing that cinnamaldehyde could also decrease the atherosclerosis plaques through TRPA1 activation. It was further shown that TRPA1-mediated Ca2+ entry reduced macrophage transition toward the inflammatory M1 phenotype while it favored M2 polarization (Wang et al., 2020; Wang et al., 2023a), thereby contributing to activating anti-inflammatory and tissue repair responses (Perez and Rius-Perez, 2022). Therefore, these findings strongly suggest that TRPA1 activation represents a promising strategy for anti-atherosclerotic treatments (Gao et al., 2020).
TRPA1 in HypPrec
An intimate relationship between hypoxia and TRPA1 has recently been established (Figure 3) (Takahashi et al., 2011). Curiously, TRPA1 is not only sensitive to hypoxia but also to hyperoxia (Takahashi et al., 2011), which is also a crucial feature of HIF signaling (see above).
Association of hypoxia with TRPA1
TRPA1 expression is regulated by HIF-1α (Figure 3) (Hatano et al., 2012). In accord, the TRPA1 gene contains a specific hypoxia response element-like motif that can be bound by HIF-1α, which can increase TRPA1 expression (Hatano et al., 2012). Moreover, both HIF-1α and TRPA1 proteins can be regulated by the hypoxia-sensing prolyl hydroxylases (PHDs) (Mori et al., 2017; Talavera et al., 2020). Under normoxic conditions (~20% O2), PHD tonically inhibit TRPA1 activity via hydroxylation of Pro394, which is located on the NH2-terminal ARD domain of the channel protein (Takahashi et al., 2011). A reduction in PO2, therefore, relieves TRPA1 from PHD-dependent inhibition and stimulates TRPA1-mediated Ca2+ entry under hypoxia (Takahashi et al., 2011). The consensus sequence for PHD-dependent hydroxylation flanking the Pro394 residue in TRPA1 protein is the same as that present in the prolyl hydroxylation motif of HIF-1 and HIF-2 (Takahashi et al., 2011; Mori et al., 2017). In addition, hypoxia can stimulate the insertion of non-hydroxylated TRPA1 channel proteins on the plasma membrane, thereby further enhancing TRPA1-mediated Ca2+ entry (Takahashi et al., 2011). Currently, TRPA1 is regarded as a molecular sensor of local %O2 changes (Mori et al., 2017; Talavera et al., 2020). Therefore, hypoxia can potentially stimulate TRPA1 activation in the cardiovascular system and could play a role in HypPrec. In the next paragraph, we discuss the evidence that appropriate TRPA1 activation by hypoxia could recruit cardioprotective Ca2+-dependent pathways.
The role of hypoxia-induced TRPA1 activation in the cardiovascular system
Cerebrovascular endothelial cells are enriched with TRPA1, which is sensitive to ROS produced during neuronal activity and triggers an increase in local cerebral blood flow to match oxygen and nutrient perfusion with the increased neuronal demand (Alvarado et al., 2021; Thakore et al., 2021). Pires and Earley (2018) demonstrated that hypoxia stimulated TRPA1-mediated Ca2+ entry in the endothelial cells lining of mouse cerebral arteries, as also reviewed in (Alvarado et al., 2021). TRPA1-mediated Ca2+ signals mediated hypoxia-induced vasodilation of cerebral arteries by recruiting IKCa/SKCa channels and promoting EDH. Consistent with these findings, cerebral damage after brain ischemia was significantly enhanced in TRPA1 knockout mice. Furthermore, the pharmacological activation of TRPA1 after the induction of brain ischemia reduced the infarct size and proved to be neuroprotective (Pires and Earley, 2018). Although future studies could compare the neuroprotective effect of TRPA1 activation prior to brain ischemia, these pieces of evidence strongly suggest that endothelial TRPA1 activity can reduce cerebral injury associated with brain stroke.
Hypoxia could also lead to TRPA1 activation in cardiomyocytes. Liu et al. (2021) showed that the pharmacological and genetic blockade of PHD2 activity resulted in TRPA1-mediated Ca2+ entry in cardiomyocytes, followed by sarcoplasmic reticulum Ca2+ release via Ca2+-induced Ca2+ release through ryanodine receptors. TRPA1-mediated Ca2+ signals activated both CaMKII and adenosine monophosphate (AMP)-dependent protein kinase (AMPK) (Liu et al., 2021). CaMKII can enhance cardiac contractility, as discussed above, whereas AMPK can rescue energy metabolism and promote cell survival during hypoxia (Naryzhnaya et al., 2020; Popov et al., 2023). Therefore, these findings strongly suggest that TRPA1 activation during hypoxia can exert a cardioprotective effect on the heart.
The role of TRPA1 signaling in HypPrec: current perspectives
TRPA1 is emerging as a critical molecular tool to translate short hypoxic bursts into a cardioprotective signal. Future work is needed to solve the controversy regarding the ability of TRPA1 to modulate cardiac fibrosis and to assess whether TRPA1 activation in coronary endothelial cells may induce vasodilation and, possibly, post-ischemic neo-angiogenesis. Nevertheless, available evidence highlights TRPA1-based mechanisms as key drivers of the HypPrec process. In agreement with these observations, other members of the TRP superfamily have been shown to protect the cardiovascular system from ischemic insults (Randhawa and Jaggi, 2015). For instance, the ability of the TRPV1 pathway to induce cerebral IschPrec (Thushara Vijayakumar et al., 2016) as well as HypPrec in rat hearts (Lu et al., 2014) has been elucidated. Furthermore, TRPV1 could also activate cardioprotective pathways during ischemic post-conditioning and remote ischemic post-conditioning (Randhawa and Jaggi, 2017). Vascular HypPrec also relies on TRPV4-dependent Ca2+influx and proper intercellular gap junctions’ communication (Rath et al., 2012). TRP-based mechanisms are also involved in sevoflurane-mediated preconditioning, which promotes mesenchymal stem cells to relieve myocardial ischemia/reperfusion injury via TRP canonical 6 (TRPC6)-induced angiogenesis via HIF-1α, C-X-C chemokine receptor type 4, and vascular endothelial growth factor (Yang et al., 2021). It is worth pointing out that several dietary and synthetic TRPA1 agonists are now available that could pharmacologically exploit the activation of TRPA1 for cardioprotective purposes (Moccia et al., 2019; Talavera et al., 2020; Thakore et al., 2020; Alvarado et al., 2021). TRPA1 signaling is likely to play a critical role during HypPrec. Confirming TRPA1 contribution could be instrumental in designing novel therapeutic approaches for preconditioning medicine. The cardioprotective effects of TRPA1 activation by the photosensitive agonist, optovin, shed novel light on the pharmacological manipulation of TRPA1 (Kokel et al., 2013; Lam et al., 2017). Optical stimulation of the target proteins offers an unprecedented spatio-temporal resolution to rescue the injured functions of a specific organ target (Di Maria et al., 2018; Moccia et al., 2022b; Moccia et al., 2023b; Vurro et al., 2023). Being itself photosensitive, optovin does not require the genetic manipulation of host cells, as required by optogenetics and chemogenetics (Di Maria et al., 2018). Unfortunately, optovin is sensitive to relatively short wavelengths (~400 nm) that do not effectively penetrate tissue and are, therefore, not easily exploitable for therapeutic purposes. Nevertheless, a number of photosensitive nanomaterials, which are highly conformable, biocompatible, and suitable for in vivo applications, have recently been developed (Zucchetti et al., 2017; Di Maria et al., 2018; Antognazza et al., 2019; Aziz and Antognazza, 2020). These nanomaterials, including regioregular Poly (3-hexyl-thiophene) (P3HT) (Lodola et al., 2019b; Moccia et al., 2022b; Negri et al., 2022; Criado-Gonzalez et al., 2023), poly[2,1,3-benzothiadiazole-4,7-diyl[4,4-bis(2-ethylhexyl)-4H-cyclopenta[2,1-b:3,4-b']dithiophene-2,6-diyl]] (PCPDTBT) (Ronchi et al., 2024), poly[(2,6-(4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)-benzo[1,2-b:4,5-b′]dithiophene))-alt-(5,5-(1′,3′-di-2-thienyl-5′,7′-bis(2-ethylhexyl)benzo[1′,2′-c:4′,5′-c′]dithiophene-4,8-dione)] (PBDB-T), and poly[[4,8-bis[(2-ethylhexyl)oxy]benzo[1,2-b:4,5-b′]dithiophene-2,6-diyl][3-fluoro-2-[(2-ethylhexyl)carbonyl]thieno[3,4-b]thiophenediyl]] (PTB7) (Tullii et al., 2023), are sensitive to longer wavelengths and generate ROS in response to light. It has been demonstrated that optical stimulation of photosensitive conjugated polymers, in the form of either thin films or nanoparticles, can stimulate angiogenesis and cardiac contraction through a mechanism that involves both ROS and TRPV1-mediated Ca2+ entry (Lodola et al., 2019a; Negri et al., 2020; Moccia et al., 2022b). However, this same signaling pathway has also been proposed to activate TRPA1 (Negri et al., 2020; Moccia et al., 2022b). Therefore, future studies that assess whether the photostimulation of conjugated polymers also results in cardioprotective effects upon TRPA1 activation would provide therapeutically relevant results.
Conclusions
TRPA1 signaling is likely to play a critical role during HypPrec. Confirming TRPA1 contribution could be instrumental in designing novel therapeutic approaches for preconditioning medicine. A critical step is to determine how TRPA1-mediated Ca2+ entry protects cardiomyocytes from cell death. As outlined above, multiple mechanisms can contribute to cardiac injury or reperfusion, and although all of them ultimately lead to cell death, some are more prone to induce an inflammatory response than others (e.g., pyroptosis vs. necrosis and apoptosis) (Davidson et al., 2019; Davidson et al., 2020). The current evidence shows that TRPA1-mediated Ca2+ entry may inhibit pyroptosis (Wang et al., 2023a). Moreover, in some cancer cell types, TRPA1 is coupled with an antioxidant system that mitigates the oxidative stress and the mitochondrial Ca2+ overload associated with apoptosis (Moccia and Montagna, 2023). Therefore, the cardiac TRPA1 could engage multifaceted signaling pathways that protect cardiomyocytes from various regulated cell death processes. The therapeutic translation of TRPA1 for HypPrec strategies also requires the elucidation of its functional role in coronary VSMCs (if present) and endothelial cells (Davidson et al., 2019), as well as in pericytes (if present) (Frangogiannis, 2024), which regulate coronary flow and permeability. Being expressed in multiple cellular compartments of the cardiovascular system, TRPA1 could provide an ideal molecular target to activate the multitarget strategy that has recently been proposed as the most suitable approach to reduce myocardial injury (Davidson et al., 2019).
Conflicts of interest
The authors declare that they have no conflicts of interest.
Acknowledgments
None.
References
Francesco Moccia1
1Department of Medicine and Health Sciences “V. Tiberio”, University of Molise, 86100 Campobasso.
Michele Samaja2
2Department of Health Science, University of Milan, 20142 Milan, Italy.
Corresponding author:
Francesco Moccia
Email: francesco.moccia@unimol.it
or
Michele Samaja
Email: michele.samaja@unimi.it
In a new window | Download PPT
Figure 1. TRPA1-mediated Ca2+ entry recruits Ca2+-dependent vasorelaxant pathways in proximity to the vascular wall. Extracellular Ca2+ entry through TRPA1 can stimulate eNOS activity, resulting in robust NO release. In addition, TRPA1-mediated Ca2+ signals can activate the small- and intermediated-conductance Ca2+-dependent K+ channels, SKCa and IKCa, which causes endothelium-dependent hyperpolarization (EDH). EDH is then electrotonically propagated to adjacent vascular smooth muscle cells (VSMCs) through myo-endothelial gap junctions, thereby causing VSMC hyperpolarization and relaxation. An alternative mechanism by which TRPA1 activation can induce vasorelaxation is by stimulating the release of NO and CGRP (as well as substance P) from the sensory fibers in close contact with resistance vessels.
In a new window | Download PPT
Figure 2. TRPA1-mediated Ca2+ entry stimulates Ca2+-dependent cardioprotective pathways in cardiac myocytes. TRPA1-mediated Ca2+ entry stimulates CaMKII, which in turn phosphorylates sarcoendoplasmic reticulum Ca2+-ATPase 2a (SERCA) and type 2 ryanodine receptors (RyR2) to, respectively, sequester cytosolic Ca2+ more avidly (lusitropic effect) and release sarcoplasmic reticulum (SR) Ca2+ more efficiently (inotropic effect). In addition, TRPA1-mediated Ca2+ signals stimulate NO release by inducing Akt-dependent eNOS phosphorylation and, possibly, via direct eNOS activation.
In a new window | Download PPT
Figure 3. Hypoxia regulates TRPA1 signalling. Under normoxic conditions, PHDs hydroxylate Pro354 on the NH2-terminal ARD domain of the TRPA1 protein, thereby inhibiting the channel and preventing basal Ca2+ entry. The drop in local %O2 under hypoxia inhibits PHD activity, which results in Pro354 dehydroxylation and TRPA1 activation. In addition, hypoxia can lead to the insertion of non-hydroxylated TRPA1 proteins on the plasma membrane, which also results in the increase of basal TRPA1-mediated Ca2+ entry. Finally, hypoxia can increase the expression of TRPA1 protein as the TRPA1 gene contains a hypoxia-sensitive element that is bound by HIF-1.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 11920 | 16 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA