International bi-monthly journal of cell signaling, tissue protection, and translational research.
Mitochondrial hexokinase as end-effector of cardioprotection
Author Affiliations

Abstract
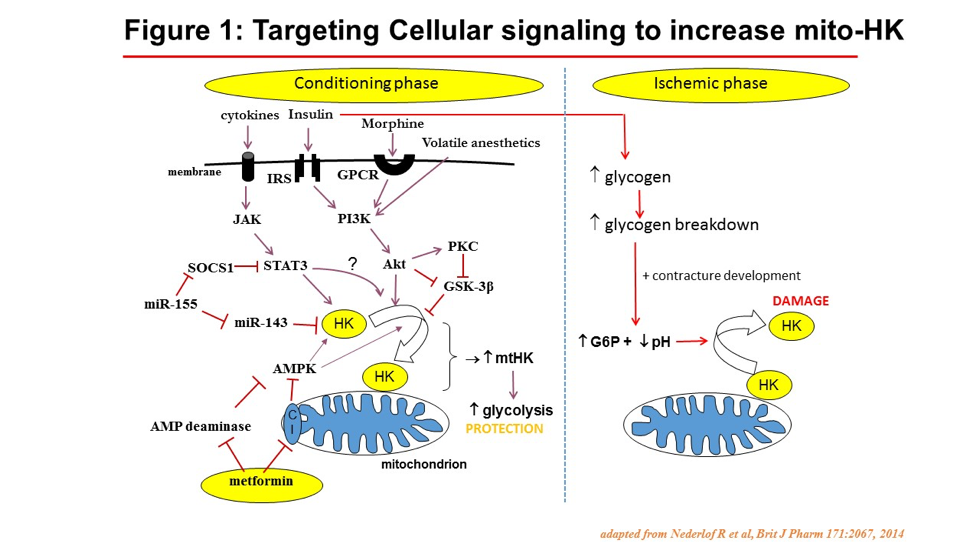
Here we summarize previous and new data from several reseach groups, identifying the crucial role of the association of the glycolytic enzyme hexokinase II (HKII) to the mitochondria for protecting the cell against cell death. Previous research demonstrated that only when glucose is present, ischemic preconditioning (IPC) protects ànd slows down mitochondria, showing that IPC works through glycolysis and mitochondria. This interaction between glycolysis and mitochondria is to a large part facilitated by increased binding of HKII to mitochondria (mitoHKII). The best ischemia-protected cells are tumor cells, with > 100 x increased HK expression as normal cells, thereby also enabling the clinical diagnosis of these tumor cells through the use of fluo-deoxyglucose imaging (FDG Spect). Several cardioprotective interventions result in increased mitoHKII in the heart. Detaching HKII from mitochondria with custom-made TAT peptides abolished IPC. Acutely detaching large amounts of HKII from mitochondria almost completely destroys the heart within 15 min, showing severely swollen mitochondria. Decreasing mitoHKII lowers mitochondrial potential, enhances ROS production at reperfusion, and turns reversible ischemia (15 min of ischemia) into irreversible, damaging ischemia of the intact heart. An inverse lineair relation exists between infartc size and mitoHKII. During ischemia, the build up of glucose-6-phosphate and increased acidosis, both from glycogen breakdown, cause detachment of HKII from mitochondria. Therefore, insulin-perfused isolated hearts experience more I/R injury than non-inuslin perfused hearts, due to increased cardiac glycogen at start of ischemia. It has been suggested that the detachment of HKII from mitochondria is the primal initial event causing mPTP opening, only later followed by ROS production. Alterations in mtHKII can be considered as end-effector mechanism of various compounds/signalling pathways that have been associated with protection (melatonin, HIF-1a, opiods, volatile anesthetics, insulin, cytokines, JAK/STAT3, PI3/Akt, GSK3β, AMPK; Fig. 1). The quest is now to develop therapy to instantanously increase or maintain mitoHKII during ischemia and early reperfusion.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 737 | 0 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA