Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Targeting metabolism to treat heart failure
Time:2023-12-01
Number:8130
Author Affiliations

Conditioning Medicine 2023. 6(2): 54-64.
Abstract
Heart failure (HF) is a complex and progressive problem characterized by the heart's inability to adequately supply the body with oxygen and nutrients. It poses a significant burden on global healthcare systems, particularly in high-income countries. This review focuses on the potential and emerging metabolic targets and therapies for treating HF and improving patients' quality of life. It explores the differences in energy-producing mitochondrial and metabolic pathways between normal individuals and HF patients. By understanding the alterations in energy metabolism associated with HF, valuable insights can be gained for the development of targeted therapies that can restore energy balance and improve cardiac function such as increasing myocardial ketone uptake, stimulating glucose oxidation, improving cardiac insulin signaling, alternation in fatty acid oxidation, and stimulating branched-chain amino acid (BCAA) oxidation. Targeting metabolism offers promising avenues for the treatment of HF and holds potential for addressing the unmet medical needs of this condition.
Keywords: Heart failure, Metabolism, Fatty acid oxidation, Glycolysis, Glucose oxidation, Ketone body oxidation
Abstract
Heart failure (HF) is a complex and progressive problem characterized by the heart's inability to adequately supply the body with oxygen and nutrients. It poses a significant burden on global healthcare systems, particularly in high-income countries. This review focuses on the potential and emerging metabolic targets and therapies for treating HF and improving patients' quality of life. It explores the differences in energy-producing mitochondrial and metabolic pathways between normal individuals and HF patients. By understanding the alterations in energy metabolism associated with HF, valuable insights can be gained for the development of targeted therapies that can restore energy balance and improve cardiac function such as increasing myocardial ketone uptake, stimulating glucose oxidation, improving cardiac insulin signaling, alternation in fatty acid oxidation, and stimulating branched-chain amino acid (BCAA) oxidation. Targeting metabolism offers promising avenues for the treatment of HF and holds potential for addressing the unmet medical needs of this condition.
Keywords: Heart failure, Metabolism, Fatty acid oxidation, Glycolysis, Glucose oxidation, Ketone body oxidation
Highlights
The heart has been the key organ target for conditioning medicine. This review explores the potential of metabolic pathways including myocardial ketone uptake, glucose oxidation, cardiac insulin signaling, fatty acid oxidation, and branched-chain amino acid oxidation as targets for enhancing energy-producing mitochondrial and metabolic activities to improve cardiac function via conditioning medicine-based therapies. This review paper presents recent progress in our understanding of metabolism in advancing conditioning medicine for treating heart failure.
Introduction
Heart failure (HF) is a serious health issue affecting more than 60 million people worldwide, and has a considerable effect at the individual and community level (James et al., 2018). Currently, 920,000 individuals in the United Kingdom are living with HF. The prevalence of HF is rising and is about 1- 3% in adults. This rise is expected to continue specifically among the elderly, due to improved diagnostics and treatments, and this prevalence varies by region and type (Savarese et al., 2023).
The incidence rates are stable or declining over time, and it differs according to geography, population, and age. Studies showed that HF incidence increases with age, and HF is more likely to develop in people at lower socioeconomic levels (Conrad et al., 2018). There are many causes of HF, with ischemic heart disease (IHD) being the most prevalent cause, accounting for almost 40% of the HF cases worldwide (Vedin et al., 2017). Other significant causes include hypertension, rheumatic heart disease (RHD), and Chagas cardiomyopathy (Fonarow et al., 2007; de Andrade et al., 2011; Watkins et al., 2017). Furthermore, there are other potential etiologies, but the available data are limited. Despite advancements in medical care and prognosis, treatments for HF remain limited. HF patients continue to face high mortality rates, with a 5-year risk of up to 75% in some demographics, frequent hospitalizations, and cardiovascular and non-cardiovascular comorbidities impacting outcomes (Savarese et al., 2023). According to one study, HF presents a significantly higher burden on the National Health Service (NHS) when compared to the combined burden of the four most common cancers (Stewart et al., 2001). The economic burden of HF is also substantial. For example, it accounts for £625 million of NHS costs, which corresponds to approximately 1-2% of the annual budget (Braunschweig et al., 2011). Symptoms like shortness of breath, weariness, fluid retention, and inability to exercise can be considered typical symptoms (Bozkurt et al., 2021). Physical examination findings can include elevated jugular venous pressure and heart murmur. Additionally, HF may present with specific signs such as hepatojugular reflux, S3 gallop, and murmurs indicative of valvular regurgitation. Diagnosis of HF typically involves a physical examination, medical history, blood tests, electrocardiogram, and imaging tests such as X-rays and echocardiography. While the normal heart is capable of efficiently utilizing various energy sources to generate adenosine triphosphate (ATP) and sustain its continuous contractions, alterations in energy-producing mitochondrial and metabolic pathways occur in HF. These alterations, including mitochondrial dysfunction and reduced oxidative metabolism, lead to a loss of flexibility in energy substrate utilization. Consequently, the failing heart undergoes a shift from fatty acid metabolism to glucose metabolism in an attempt to sustain ATP production. Understanding these changes in energy metabolism is crucial for exploring potential therapeutic interventions.
Energy-Producing Mitochondrial and Metabolic Pathways in Normal Hear
The heart's need for energy is very high due to the continuous and rhythmic contractions, and to support its function, constant ATP generation must occur (Lopaschuk et al., 2010; Karwi et al., 2018). This ATP production is accomplished through energy-producing mitochondrial and metabolic pathways, which include oxidative phosphorylation (95%), glycolysis (5%), and citric acid cycle, where different fuel sources such as fatty acids, glucose, lactate, ketones, and amino acids are converted into ATP (Figure 1) (Wisneski et al., 1990; Saddik and Lopaschuk, 1991). Most of this ATP is used by the heart for contraction, and the remaining ATP is used for ion pumps (Gibbs, 1978; Suga, 1990). The normal heart is flexible and can quickly switch between several energy sources to sustain ATP generation (Karwi et al., 2018).
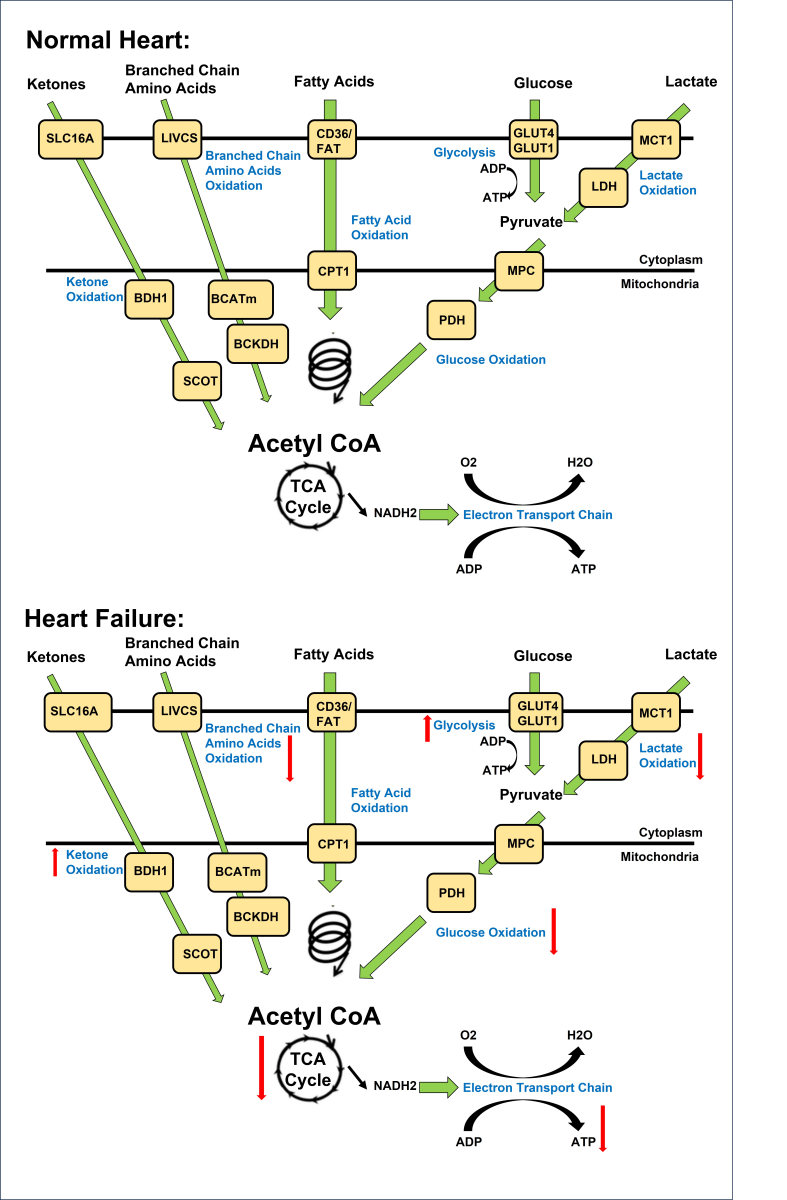
In a new window | Download PPT
Figure 1. Overview of energy metabolism in the normal and failing heart. Normal Heart: Glucose is transported into the cell via GLUT1 or GLUT4 and undergoes glycolysis, producing pyruvate. Lactate is taken up through MCT and converted to pyruvate by LDH. Pyruvate from glucose and lactate enters the mitochondria via MPC and is converted to acetyl CoA by PDH. Fatty acids are transported into cardiomyocytes through CD36 and FATP-1, where they are esterified to fatty acyl-CoA. The acyl group is transferred to carnitine by CPT-1 and transported into the mitochondria. CPT-2 converts it back to fatty acyl CoA, which undergoes β-oxidation, producing acetyl CoA. Ketones, such as β-hydroxybutyrate, are transported into the cell through SLC16A1. BDH1 catalyzes the oxidation of β-hydroxybutyrate to acetoacetate, which is activated by SCOT to acetoacetyl-CoA. Thiolysis reaction generates acetyl CoA. BCAAs enter the cell via LIVCS and are converted to ketoacids by BCATm. Acetyl CoA and succinyl CoA are formed from BCKDH. Acetyl CoA from fatty acid β-oxidation, glucose oxidation, ketone oxidation, and BCAA oxidation enters the TCA cycle, generating FADH2 and NADH. These molecules then enter the electron transport chain, consuming oxygen to produce ATP. Failing Heart: Changes in the oxidation of ketones, amino acids, fatty acids, glycolysis, glucose, and lactate metabolism occur in the heart during heart failure. Upward arrows signify an increase, while downward arrows indicate a decrease in these metabolic processes. Abbreviations: Glucose transporter 1 and 4 (GLUT1, GLUT4), mitochondrial pyruvate carrier (MPC), pyruvate dehydrogenase (PDH), monocarboxylate transporter (MCT), lactate dehydrogenase (LDH), CD36/fatty acid transporter (CD36/FAT), carnitine palmitoyl transferase (CPT-1), nicotinamide adenine dinucleotide (NADH2), adenosine triphosphate (ATP), adenosine diphosphate (ADP), tricarboxylic acid (TCA), β-hydroxybutyrate (βOHβ), monocarboxylate transporter 1 (SLC16A1), β-hydroxybutyrate dehydrogenase 1 (BDH1), succinyl-CoA:3 oxoacid-CoA transferase (SCOT), branched chain amino acid cation symporter (LIVCS), mitochondrial branched chain amino-transaminase (BCATm), branched chain α-keto acid dehydrogenase (BCKDH). Adapted from (Lopaschuk et al., 2021).
Fatty Acid oxidation in normal heart
Fatty acid transport protein (FATP) and CD36 facilitate the transportation of fatty acids into cardiac muscle cells across the cell membrane. Then, through fatty acid esterification, fatty acyl-coenzyme A (CoA) is formed by adding the CoA molecule to the fatty acid (Fillmore et al., 2014). Fatty acyl-CoA is then carried into the mitochondria by carnitine palmitoyl transferase-1 (CPT-1), and subsequently CPT-2 transforms it back into fatty acyl-CoA (Murthy and Pande, 1987). Inside the mitochondria and after a series of reactions, fatty acid oxidation produces acetyl-CoA, Nicotinamide adenine dinucleotide (NADH ), and Flavin adenine dinucleotide (FADH2), which enter the citric acid cycle to produce ATP (Saddik et al., 1993).
Glucose and lactate oxidation in normal heart
Glucose is also used to produce ATP, and this process is known as glucose metabolism or glucose oxidation. The transportation of glucose into the cardiac cell is facilitated by glucose transporter 1 (GLUT1) or GLUT4 (Aerni-Flessner et al., 2012). Then through glycolysis, which happens in the cytoplasm, it is converted into two molecules of pyruvate. An additional energy source for the heart is lactate which is used through the lactate oxidation process. Lactate enters cells via the monocarboxylate transporter (MCT), and then it is converted into pyruvate by transferring electrons and protons from lactate to NAD+ (Van Hall, 2010). This chemical process is catalyzed by lactate dehydrogenase (LDH). The mitochondrial pyruvate carrier (MPC) carries the pyruvate molecules created from lactate and glucose into the mitochondria, and then these pyruvate molecules are decarboxylated by losing one carbon dioxide (CO2) molecule to form acetaldehyde, which is then oxidized to acetate. The combination of acetate with coenzyme A (CoA) forms acetyl-CoA.
Ketone oxidation in normal heart
SLC16A1 eases the transportation of ketones such as beta-hydroxybutyrate (βOHB) across the cell membrane. After entering cardiomyocytes, β-hydroxybutyrate (βOHB) is converted into acetoacetate (AcAc) via an enzyme called βOHB dehydrogenase 1 (BDH1), and this AcAc is activated by succinyl-CoA:3 oxoacid-CoA transferase (SCOT) and converted into acetoacetyl-CoA (AcAc-CoA), which passes through a thiolysis process and generates acetyl-CoA (Ho et al., 2021).
BSAAs in normal heart
Branched-chain amino acids (BCAAs) generate acetyl-CoA through a catabolism process, where BCAAs are transformed to keto acids by mitochondrial branched-chain aminotransferase (BCATm) and form branched-chain keto acids (BCKAs), and this happens after transporting the BCAAs to cells via the branched chain amino acid:cation symporter family (LIVCS). Then, branch chain acid alpha-keto acid dehydrogenase (BCKDH) uses BCKAs to create succinyl CoA and acetyl CoA. Branch-chain -keto acid dehydrogenase kinase (BCKDK) phosphorylates and inactivates BCKDH, whereas mitochondrial protein phosphatase 2C dephosphorylates and activates BCKDH (PP2Cm) (Fillmore et al., 2018).
TCA cycle and ATP synthase
Acetyl-CoA derived from fatty acids, glucose, ketones, lactate, and BCAAs enters the tricarboxylic acid (TCA) cycle, which is also called the citric acid cycle or Krebs cycle (Schwarzer and Doenst, 2015). It is a critical element of cellular respiration, where cells produce ATP through a sequence of chemical processes that take place in the cardiomyocytes' mitochondria.The enzyme citrate synthase facilitates the combination of acetyl-CoA and oxaloacetate to produce citrate. The consecutive set of enzyme-catalyzed reactions converts citrate into isocitrate, α-ketoglutarate, succinyl-CoA, succinate, fumarate, and malate (Vujic et al., 2021). These processes and reactions generate reducing equivalents, energy-rich molecules, and important intermediates, which are critical for producing ATP. For example, α-ketoglutarate produces electron carriers in the form of NADH and FADH2. Also, when succinyl-CoA is converted to succinate, ATP is directly produced. Additionally, malate is oxidized to produce NADH (Vujic et al., 2021). These NADH and FADH2 provide electrons to the electron transport chain (ETC), and these electrons enter the ETC at the first protein complex in the chain usually NADH dehydrogenase (Complex I). Then, these electrons are exchanged between several protein complexes along the chain, creating the proton gradient by releasing and using energy to pump protons (H+) to the intermembrane space. The energy from this proton gradient is used to create ATP, where these protons pass via ATP synthase and return to the mitochondrial matrix, which acts like a turbine (Vujic et al., 2021). This flow leads to the creation of ATP from ADP and inorganic phosphate (Pi), and this process is named oxidative phosphorylation (Schwarzer and Doenst, 2015). Overall, constant ATP production is vital for the heart to function effectively. ATP is produced through various metabolic pathways which are well-described and well-studied, and these pathways are the keystone of developing new metabolic targets and therapies for treating HF.
Alterations in Energy-Producing Mitochondrial and Metabolic Pathways in HF
HF is linked with mitochondrial dysfunction, which causes changes in energy metabolism and a reduction in oxidative metabolism, where the failing heart loses its flexibility to switch between energy sources to generate ATP constantly (Neubauer et al., 1997; Stanley et al., 2005). Therefore, the substrate switches from fatty acids to glucose in HF [Figure.1] (Casademont and Miró, 2002). On the other hand, as HF progresses, glucose oxidation is likewise decreased, and the heart begins to depend more heavily on other sources like ketone and lactate metabolism, and experiences increased proteolysis (Aubert et al., 2016; Bedi et al., 2016; Horton et al., 2019; Murashige et al., 2020). Previous research has indicated ATP levels in failing hearts are reduced by around 30% (Ingwall and Weiss, 2004). Numerous studies also suggested a connection between mitochondrial dysfunction in HF and post-translational changes in mitochondrial proteins and in changes mitochondrial dynamics, such as increased fission and mitophagy, repression of peroxisome proliferator-activated receptor-gamma coactivator-1 alpha, a key transcriptional regulator of mitochondrial biogenesis, and repression of peroxisome proliferator-activated receptor, which controls fatty acid oxidation (Finck and Kelly, 2006; Lopaschuk et al., 2010; Dirks-Naylor et al., 2014; Chaanine et al., 2019; Bauer and Murphy, 2020; O’Rourke et al., 2021). Cardiac insulin resistance may operate as a mediator and a possible marker of contractile dysfunction in HF.
Fattyacid oxidation and HF
A previous study suggested that the uptake and oxidation of fatty acids are reduced with the development of HF, likely due to impaired mitochondrial function and decreased activity of key enzymes involved in fatty acid oxidation (Barger and Kelly, 1999). A study by Chandler et al. (2004) suggested that these alterations in fatty acid oxidation might be influenced by the severity and phase of HF, as demonstrated by a progressive decline in fatty acid oxidation rates over time. Specifically, many studies have shown that during the initial phases of HF, there is either no change or a slight increase in fatty acid oxidation, likely due to compensatory upregulation of fatty acid transporters and beta-oxidation enzymes. However, a significant reduction in fatty acid oxidation occurs when HF developed (Chandler et al., 2004; Stanley et al., 2005). On the other hand, some studies have shown an increase in fatty acid uptake and oxidation in individuals with comorbid conditions such as congestive HF, type-2 diabetes, obesity, and insulin resistance. This response may be due to altered insulin signaling (through insulin resistance, impaired insulin secretion, or dysregulated insulin signaling pathways) and increased expression of fatty acid transporters (through hormonal imbalances, altered gene expression, or cellular adaptations) (Peterson et al., 2004; Rijzewijk et al., 2009; Voros et al., 2018). Furthermore, there have been instances where increased fatty acid oxidation has proven advantageous in HF. A recent publication (Watson et al., 2023) demonstrated that during Intralipid infusion, cardiac long-chain fatty acid uptake and oxidation increased, leading to improved myocardial energetics and contractility. Similarly, early work has highlighted how a high-fat diet increased certain cardiac metabolic activities but failed to normalize mRNA or protein levels of acyl-CoA dehydrogenases in HF rats (Rennison et al., 2008). These results are not consistent, which indicates a gap in this area due to its complexity and the alternations in other pathways. However, fatty acids still contribute more to mitochondrial ATP production in HF than glucose (Murashige et al., 2020).
Glucose and lactate oxidation and HF
The switch from oxidative phosphorylation to glycolysis is regulated by a complex network of signaling pathways, and involves the upregulation of key enzymes and transporters. For example, studies have shown that in response to HF, the expression of GLUT1 is increased, facilitating glucose uptake into the cell. Similarly, the activity of the glycolysis enzyme phosphofructokinase 1 (PFK-1) is enhanced, leading to higher glycolytic flux (Diakos et al., 2016). However, while this corrective response may help to maintain ATP levels in the short term, it is inadequate to fully compensate for the energy deficit due to the small proportion of ATP that is produced by glycolysis. Glucose oxidation is consistently decreased in HF patients. For instance, a study on pacing-induced HF in pigs showed a reduction in glucose oxidation, while similar results were observed in mouse models of HF and human patients with end-stage HF (Zhabyeyev et al., 2013; Zhang et al., 2013). In myocardial biopsies obtained from HF patients, there is a significant downregulation of key enzymes and transporters involved in pyruvate metabolism, including MCT1, pyruvate dehydrogenase complex, MPCs, and pyruvate/alanine aminotransferases. The reduced expression of these molecules suggests a decrease in pyruvate transport and metabolism in the failing heart (Doehner et al., 2014; Fernandez-Caggiano et al., 2020; Lee et al., 2020). Notably, studies on patients with HF associated with obesity, insulin resistance, and diabetes have shown a greater decrease in glucose oxidation compared to HF patients without these comorbidities (Thakker et al., 2008; Mori et al., 2012; Holzem et al., 2015). Overall, targeting the enzymes and transporters involved in pyruvate metabolism may be a promising strategy for developing new treatments for HF.
Ketones oxidation and HF
Several investigations have demonstrated that HF causes rise in ketone oxidation (Aubert et al., 2016; Bedi et al., 2016; Ho et al., 2019). For instance, one research study showed that individuals with HF with reduced ejection fraction (HFrEF) had ketone oxidation that was almost 100% higher (Funada et al., 2009). Overall, further study is required to determine how the metabolism of ketone affects the onset of HF and to compare its effectiveness to that of other substrates in the failing heart. In conclusion, alterations in energy-producing mitochondrial and metabolic pathways play a significant role in the development of HF. While our knowledge of the complex interplay between these pathways is growing, there is still much to learn about their individual and collective contributions to HF pathophysiology, which will provide new insights into the development of more effective therapies.
BCAAs and HF
Recent studies have shown that individuals with HF have increased levels of BCAAs in their blood. This increase may be due to a reduction in BCAA oxidation, which can lead to an accumulation of BCAA and their metabolites, including BCKAs, in the heart (Karwi and Lopaschuk, 2022). While elevated levels of BCAA in the myocardium have been linked to activation of the mTOR signaling pathway and worsening of cardiac remodeling, elevated levels of BCKA can inhibit the cardiac insulin signaling pathway and impair insulin-stimulated cardiac glucose oxidation, causing a reduction in cardiac ATP production (Karwi and Lopaschuk, 2022). This suggests that both BCAA and BCKA have a role in the pathophysiology of HF and may be potential targets for therapeutic intervention.
TCA cycle and ATP synthase in HF
Heart failure is characterized by disruptions in cellular energetics that have significant impact on vital metabolic pathways. In particular, the TCA or citric acid cycle experiences alterations in HF due to reduced ATP levels, impaired mitochondrial function, and changes in substrate utilization. This collective disruption directly affects the availability of intermediates necessary for efficient energy production and downstream metabolic processes (Lopaschuk et al., 2021; Takada et al., 2022). One prominent aspect is the disruption in NAD levels and its redox state (NAD+/NADH). HF is associated with a reduction in NAD levels or a disturbed NAD+/NADH ratio (Lopaschuk et al., 2021). This metabolic imbalance, coupled with mitochondrial dysfunction, leads to changes in substrate metabolism, including glycolysis, the TCA cycle, and oxidative phosphorylation. HF is characterized by disruptions in NAD metabolism, including altered levels and disturbed redox states. These changes contribute to mitochondrial dysfunction, impaired energy metabolism, and perturbed cellular processes (Lopaschuk et al., 2021).
Moreover, mitochondrial dysfunction and oxidative stress in HF impair the function of ATP synthase, the enzyme complex responsible for synthesizing ATP through oxidative phosphorylation (Lopaschuk et al., 2021; Takada et al., 2022). As a consequence, the capacity for energy production diminishes, critically impacting cardiac contractility and overall function. This intricate relationship further extends to disruptions in the TCA cycle, which can disrupt the supply of reducing equivalents to the ETC. This disruption, in turn, affects the proton gradient crucial for ATP synthase activity (Lopaschuk et al., 2021; Takada et al., 2022). Ultimately, these metabolic changes intricately contribute to the compromised energy production observed in HF and underscore the need for targeted interventions aimed at restoring proper energy metabolism and cardiac function in HF patients.
The Role of Imaging in Assessing Metabolism in HF
Imaging techniques are pivotal in assessing metabolism in HF and hold potential to aid in identifying patients who could benefit from targeted therapies (Seymour, 2003; Tamaki et al., 2009; Van Deursen et al., 2018; Rider et al., 2020; Burrage et al., 2022). Nuclear imaging methods like myocardial perfusion and molecular imaging are capable of detecting and quantifying the underlying pathophysiological processes in HF. They offer insights into cardiac structure, metabolism, and molecular targets, which contribute to a better understanding of HF phenotypes (Van Deursen et al., 2018). Noninvasive metabolic imaging techniques play a crucial role in revealing the metabolic status of the heart, enabling the identification of abnormalities that contribute to HF. In HF with preserved ejection fraction (HFpEF), advanced imaging methods can accurately quantify biomechanical features and provide insights into its function (Rider et al., 2020; Burrage et al., 2022). Cardiovascular magnetic resonance imaging (CMR) serves as a comprehensive tool for evaluating cardiac structure, function, and tissue characteristics in patients with suspected or confirmed HF (Tamaki et al., 2009). Echocardiography, another commonly used imaging technique, offers real-time visualization of the heart's structure and function, aiding in diagnosis, etiological assessment, and treatment guidance (Tamaki et al., 2009). In summary, various imaging techniques, including nuclear imaging, metabolic imaging, CMR, and echocardiography, play a critical role in evaluating metabolism in HF. Their ability to provide information about cardiac structure, function, metabolism, and molecular targets aids in characterizing HF phenotypes and identifying patients suitable for targeted therapies.
Potential and Emerging Metabolic Targets and Therapies for Treating HF
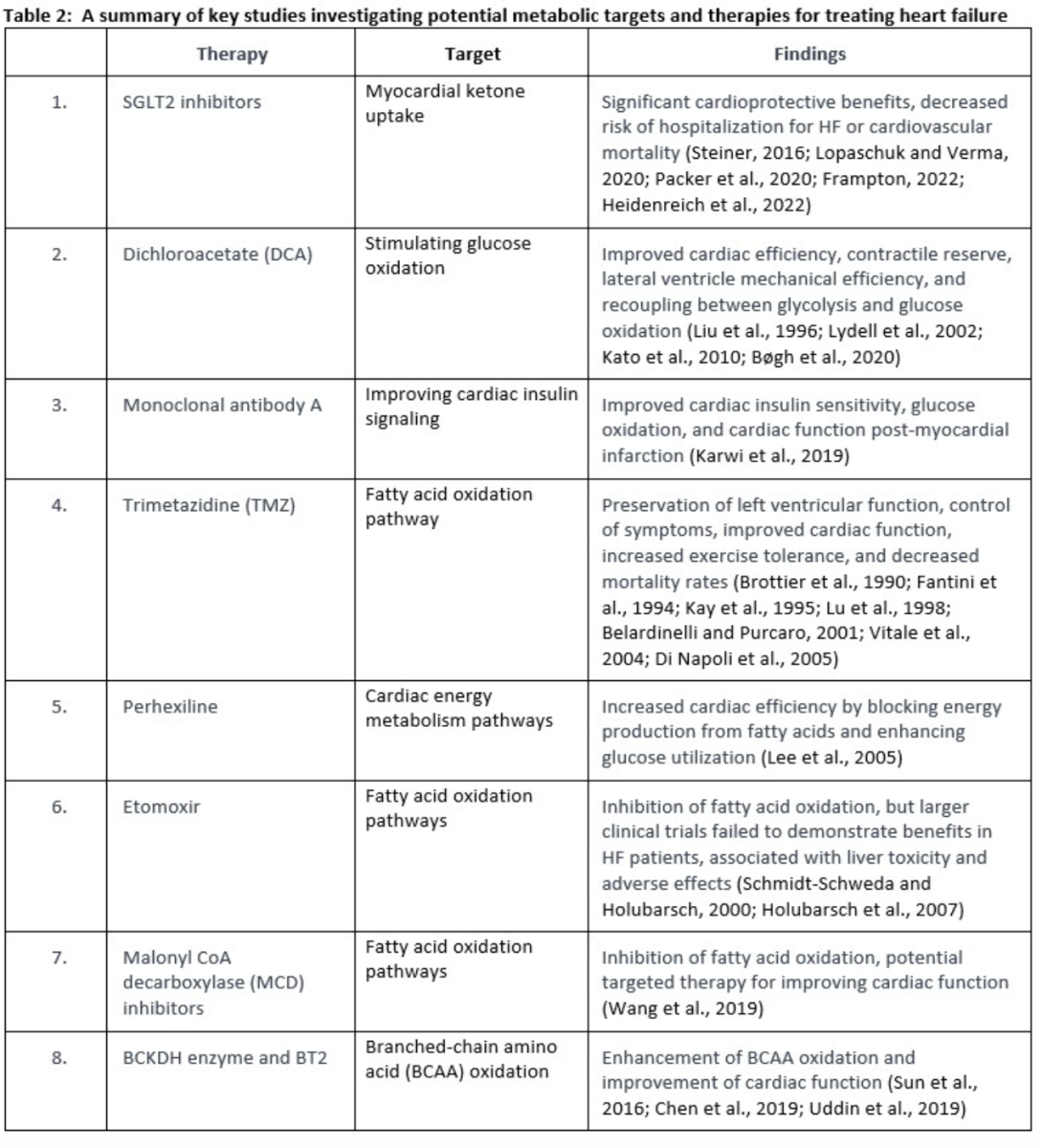
Current treatments for HF such as beta-blockers, angiotensin receptor blockers, and diuretics generally focus on reducing symptoms and enhancing cardiac function (Table.1) (McDonagh et al., 2021). While these therapies can be useful, they do not directly target the underlying metabolic and mitochondrial dysfunction. Recently, studies have proven the effects of increasing cardiac ketone oxidation by ketone infusions, sodium-glucose cotransporter-2 (SGLT2) inhibitors, or a ketogenic diet (Saucedo-Orozco et al., 2022). Other studies reported the effective use of trimetazidine as a fatty acid oxidation inhibitor, which increases glucose oxidation (Kantor et al., 2000). Furthermore, studies have proven the effectiveness of exercise to improve insulin signaling (Lopaschuk et al., 2021). On the other hand, there are many other potential and emerging therapies that may be the keystone to improving cardiac function in the future (see Table 2). These studies highlight the potential of various therapies and metabolic targets to improve cardiac function in HF. However, further research and clinical trials are needed to validate their efficacy and safety.
Increasing myocardial ketone uptake
One therapy that is included in current HF management is SGLT2 inhibitors (Heidenreich et al., 2022). SGLT2 inhibitors have recently been shown to have substantial cardioprotective benefits (Steiner, 2016; Packer et al., 2020). Increased circulating levels of ketone and increased energy delivery to the failing heart are two ways that SGLT2 inhibitors have been suggested to enhance cardiac performance in HF (Lopaschuk and Verma, 2020). Empagliflozin is one of the SGLT2 inhibitors that significantly decreased the risk of hospitalization for HF and cardiovascular mortality in nonhospitalized individuals with HFpEF, HFmrEF, or HFrEF. Additionally, empagliflozin increased the quality of life in terms of health and was usually well tolerated (Frampton, 2022).
Increasing myocardial ketone uptake is a strategy gaining attention in current HF management, notably through the utilization of SGLT2 inhibitors (Heidenreich et al., 2022). The cardioprotective benefits of SGLT2 inhibitors have been well-documented (Steiner, 2016; Packer et al., 2020), often attributed to their potential to enhance cardiac performance in HF by increasing circulating ketone levels and energy delivery to the failing heart (Lopaschuk and Verma, 2020). However, a nuanced view of this mechanism is warranted. It is essential to acknowledge that increased ketone levels with SGLT2 inhibitors are not always consistently observed, as evidenced by studies such as those by Abdurrachim et al., 2019, Kimura et al., 2019, and Hundertmark et al., 2023, where increases in ketones were modest or not present. Even when observed, the magnitude of the increase can be minimal, as indicated by a study reporting a 0.02 mM rise in ketone levels (Selvaraj et al., 2022). Intriguingly, in some cases, SGLT2 inhibitor treatment has been associated with decreased ketone oxidation (Abdurrachim et al., 2019).
Furthermore, the relationship between SGLT2 inhibitors' cardioprotective effects and ketone levels appears more complex. Contrary to the assumption that increased plasma ketones and cardiac ketone oxidation mediate the protective effects of SGLT2 inhibitors, the fact that these cardiovascular benefits are similarly effective in both diabetic and non-diabetic patients suggests a more multifaceted mechanism (Ferrannini et al., 2016). Notably, the impact of SGLT2 inhibitors on ketone levels is more pronounced in diabetic patients compared to non-diabetic individuals. These observations challenge the oversimplified link between increased ketones and cardioprotection. Therefore, while increasing myocardial ketone uptake remains a promising avenue for HF management through SGLT2 inhibitors, it is imperative to acknowledge the variability and nuances in the relationship between ketone levels, SGLT2 inhibitor therapy, and their cardioprotective effects. A comprehensive understanding of these intricacies will be pivotal in refining therapeutic strategies and optimizing HF management using SGLT2 inhibitors. Empagliflozin, for instance, has exhibited significant reduction in the risk of HF hospitalization or cardiovascular mortality across HF phenotypes (HFpEF, HFmrEF, or HFrEF) while simultaneously improving health-related quality of life (Frampton, 2022).
Stimulating glucose oxidation
One of these potential and emerging treatments is dichloroacetate, which inhibits pyruvate dehydrogenase kinase and leads to increased glucose oxidation by enhancing pyruvate dehydrogenase flux. In-vitro, in-vivo, and some clinical trial studies have shown the impact of dichloroacetate on improving cardiac efficiency, contractile reserve, lateral ventral mechanical efficiency, and recoupling between glycolysis and glucose oxidation, and these results are promising (Liu et al., 1996; Lydell et al., 2002; Kato et al., 2010; Bøgh et al., 2020).
Improving cardiac insulin signaling
Human monoclonal antibody A is another potential treatment that works as an antagonist of glucagon where it targets the G-protein coupled receptor. Inhibiting G-protein coupled receptor leads to improving insulin signaling, which improves glucose oxidation and has a positive impact on the heart. Monoclonal antibody A has shown promising results in improving cardiac insulin sensitivity, glucose oxidation, and cardiac function post-myocardial infarction (Karwi et al., 2019). Additionally, calycosin, as a PI3K/AKT activator, may have a beneficial impact on HF by reducing inflammation and fibrosis by inhibiting the AKT-IκB kinase/signal transducer and activator of transcription-3 axis. However, further studies are needed to validate these findings (Wang et al., 2022b).
Inhibiting fatty acid oxidation
Trimetazidine is a medication that exerts its effects by inhibiting oxidative phosphorylation and altering energy production by shifting energy production from free fatty acids to glucose oxidation (Fantini et al., 1994; Kay et al., 1995). Several studies indicated potential benefits of trimetazidine. These benefits include the preservation of left ventricular function and the control of symptoms associated with HF. Furthermore, trimetazidine has shown promise in improving cardiac function, reducing symptoms of HF, increasing exercise tolerance, and decreasing all-cause mortality, cardiovascular events, and hospitalization rates (Brottier et al., 1990; Lu et al., 1998; Belardinelli and Purcaro, 2001; Vitale et al., 2004; Di Napoli et al., 2005). Overall, the evidence suggests that trimetazidine can be a valuable therapeutic option for patients with HF, leading to improved outcomes and quality of life.
Perhexiline is a medication that works by blocking the production of energy from fatty acids in the heart muscle, which makes the heart use glucose more efficiently and increases cardiac efficiency (Lee et al., 2005). However, this medication has some limitations such as it can cause liver damage, peripheral neuropathy, and other serious side effects. Also, it can interact with other medications, such as antiarrhythmic drugs (Schwarzer and Doenst, 2015; Paliard et al., 1978).
Another fatty acid oxidation inhibitor is etomoxir, which inhibits the activity of CPT-1, an enzyme that transports fatty acids into the mitochondria for ATP production (Schmidt-Schweda and Holubarsch, 2000; Holubarsch et al., 2007). While etomoxir has shown promise as a therapy for HF, larger clinical trials have failed to demonstrate benefits in HF patients, it can cause liver toxicity and other adverse effects, and it inhibits the activity of CPT-1 in all tissues, not just in the heart (Wang et al., 2018). Also, malonyl CoA decarboxylase inhibitors increase the levels of malonyl CoA, a key intermediate metabolite in the fatty acid synthesis pathway, which inhibits the transport of fatty acids into the mitochondria, and can be a targeted therapy for inhibiting fatty acid oxidation and improving cardiac function (Wang et al., 2019).
Stimulating BCCA oxidation
In murine models of ischemic and failing hearts, improving the flow via the BCKDH enzyme can boost BCAA oxidation and decrease both BCAA and BCKA levels, which enhances cardiac function. The allosteric inhibitor BT2 is a successful therapy for increasing BCAA oxidation throughout the body, not only in the heart (Sun et al., 2016; Chen et al., 2019; Uddin et al., 2019).
Overall, these therapies are under review, and they have potential as promising treatment for HF.
To summarize, it's increasingly evident that a comprehensive understanding of the metabolic state of the heart is essential before attempting to alter specific pathways. The oversimplification of increasing one pathway and decreasing another without considering the intricate metabolic interplay could lead to unintended consequences. For instance, the conventional approach of increasing glucose oxidation might not be advantageous if glucose oxidation is already elevated, and a similar principle applies to other metabolic pathways. Instead of solely focusing on enhancing or inhibiting particular pathways, a more nuanced strategy could involve re-balancing metabolism. This means considering that metabolic pathways not only dictate energy production but also provide crucial metabolic intermediates that play regulatory roles within the heart. By addressing this intricate network, interventions can be aimed at restoring a harmonious equilibrium among different pathways. A study by Glatz et al. 2020 underscores the significance of this approach. It highlights that the optimal functioning of the heart depends on a balanced utilization of substrates, particularly fatty acids and glucose. During times of stress, such as in cardiac dysfunction, this balance can be disrupted, leading to an overemphasis on either fatty acids or glucose as fuel sources. This imbalance has been correlated with compromised cardiac performance. To counteract this, interventions that restore a more balanced reliance on both fatty acids and glucose have demonstrated positive outcomes in terms of cardiac function. This perspective prompts a shift from a tunnel-vision approach of targeting individual pathways to a broader perspective of harmonizing the complex web of metabolic processes. This understanding acknowledges that the metabolic intermediates generated within these pathways hold regulatory functions beyond just energy production. By re-establishing a balanced metabolic environment within the heart, the aim is to mitigate dysfunction and restore optimal cardiac performance. As research in this area continues to unfold, the concept of re-balancing metabolism emerges as a promising strategy for treating cardiac failure and promoting overall cardiac health.
Comorbidities and Cardiac Metabolism in HF
Several comorbid conditions and disturbances of heart metabolism frequently accompany HF. Understanding the interaction between these factors is crucial for developing sufficient treatments. For the development of effective therapies, it is essential to comprehend how these elements interact.
Age
Advanced age is a well-established risk factor for HF (Ma and Li, 2015). With aging, there are structural and functional changes in the heart, including alterations in cardiac metabolism. The alterations in metabolism observed in the aging heart, including impaired mitochondrial function, increased free radical production, altered glucose and fatty acid utilization, and metabolic shift from fatty acid towards glucose oxidation can collectively contribute to the development and progression of HF. These metabolic changes result in energy deficits, compromised cardiac function, and increased susceptibility to cardiac dysfunction (Nohl and Hegner, 1977; Hansford, 1983; Nohl, 1987; Kates et al., 2003).
Diabetes mellitus
In HF patients, diabetes mellitus is a frequent comorbidity that is linked to worse clinical outcomes and increase mortality and morbidity rates (Danaei et al., 2011). In diabetics, impaired insulin signaling and insulin resistance contribute to abnormal cardiac metabolism. Reduced glucose uptake and utilization in the heart as a result of insulin resistance encourages a greater dependence on fatty acid oxidation for energy generation (Peterson et al., 2004; Rijzewijk et al., 2009).
This metabolic disorder can worsen HF or hasten the development of HF by causing myocardial lipid buildup, mitochondrial dysfunction, and oxidative stress (Shah et al., 2010).
Obesity
Obesity is a significant risk factor for HF and is characterized by excessive adipose tissue accumulation. Adipose tissue produces various adipokines and inflammatory mediators that can contribute to systemic inflammation, insulin resistance, and mitochondrial dysfunction (Seidell et al., 1990; Cefalu et al., 1995; Kuk et al., 2006; Wang et al., 2022a). In obese individuals, increased circulating fatty acids and ectopic lipid deposition in the heart can lead to mitochondrial dysfunction, oxidative stress, impaired insulin signaling, and impaired cardiac metabolism (Randle et al., 1963; Kanaley et al., 2009). Obesity-induced alterations in adipokine secretion can also influence cardiac remodeling and contribute to the development and progression of HF (Borlaug et al., 2023).
Other comorbidities
Other comorbidities commonly associated with HF, such as hypertension, dyslipidemia, and chronic kidney disease, can also impact cardiac metabolism (Van Deursen et al., 2014). Overall, understanding the complex relationship between comorbidities, cardiac metabolism, and HF is essential for the development of targeted therapies. Emerging treatment strategies aim to directly modulate metabolic pathways, improve insulin signaling, and promote a shift towards more efficient cardiac energy utilization. These therapies hold promise in mitigating the impact of comorbidities on cardiac metabolism and improving outcomes in HF patients. However, further research is needed to fully elucidate the mechanisms underlying these metabolic perturbations and evaluate the effectiveness of emerging therapies in clinical settings.
Conclusion
In conclusion, HF is a complicated issue characterized by impaired cardiac function and energy production. While there have been significant advancements in understanding the role of mitochondrial and metabolic dysfunction in the development of HF, there is still much to be learned. More research is needed to fully understand the complex alterations in energy-producing pathways that occur in HF. Future research should continue to explore these potential targets and therapies, as well as new targets and therapies that may emerge, to improve the treatment of HF. Importantly, therapy should be directed at re-balancing metabolism, recognizing that all metabolic pathways also provide metabolic intermediates with important regulatory functions in the heart beyond their metabolic fate. In particular, a deeper understanding of the interplay between metabolic and mitochondrial dysfunction and cardiac function may lead to more effective therapies for HF.
Conflicts of interest
There are no conflicts of interest regarding the publication of this manuscript.
References
Ali Abdullah
Institute of Cardiovascular Science, University College London, London, UK.
Corresponding author:
Ali Abdullah
Email: ali.abdullah.22@ucl.ac.uk; Ali.nassr.abdullah@gmail.com
In a new window | Download PPT
Figure 1. Overview of energy metabolism in the normal and failing heart. Normal Heart: Glucose is transported into the cell via GLUT1 or GLUT4 and undergoes glycolysis, producing pyruvate. Lactate is taken up through MCT and converted to pyruvate by LDH. Pyruvate from glucose and lactate enters the mitochondria via MPC and is converted to acetyl CoA by PDH. Fatty acids are transported into cardiomyocytes through CD36 and FATP-1, where they are esterified to fatty acyl-CoA. The acyl group is transferred to carnitine by CPT-1 and transported into the mitochondria. CPT-2 converts it back to fatty acyl CoA, which undergoes β-oxidation, producing acetyl CoA. Ketones, such as β-hydroxybutyrate, are transported into the cell through SLC16A1. BDH1 catalyzes the oxidation of β-hydroxybutyrate to acetoacetate, which is activated by SCOT to acetoacetyl-CoA. Thiolysis reaction generates acetyl CoA. BCAAs enter the cell via LIVCS and are converted to ketoacids by BCATm. Acetyl CoA and succinyl CoA are formed from BCKDH. Acetyl CoA from fatty acid β-oxidation, glucose oxidation, ketone oxidation, and BCAA oxidation enters the TCA cycle, generating FADH2 and NADH. These molecules then enter the electron transport chain, consuming oxygen to produce ATP. Failing Heart: Changes in the oxidation of ketones, amino acids, fatty acids, glycolysis, glucose, and lactate metabolism occur in the heart during heart failure. Upward arrows signify an increase, while downward arrows indicate a decrease in these metabolic processes. Abbreviations: Glucose transporter 1 and 4 (GLUT1, GLUT4), mitochondrial pyruvate carrier (MPC), pyruvate dehydrogenase (PDH), monocarboxylate transporter (MCT), lactate dehydrogenase (LDH), CD36/fatty acid transporter (CD36/FAT), carnitine palmitoyl transferase (CPT-1), nicotinamide adenine dinucleotide (NADH2), adenosine triphosphate (ATP), adenosine diphosphate (ADP), tricarboxylic acid (TCA), β-hydroxybutyrate (βOHβ), monocarboxylate transporter 1 (SLC16A1), β-hydroxybutyrate dehydrogenase 1 (BDH1), succinyl-CoA:3 oxoacid-CoA transferase (SCOT), branched chain amino acid cation symporter (LIVCS), mitochondrial branched chain amino-transaminase (BCATm), branched chain α-keto acid dehydrogenase (BCKDH). Adapted from (Lopaschuk et al., 2021).
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 8130 | 25 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA